高效液相色譜指紋圖譜評價瀉白散及其3種單味藥內(nèi)在質(zhì)量(二)
發(fā)布時間:2021-11-14 19:42
編輯者:特邀作者周世紅
1.3 溶液制備
1.3.1 對照品溶液
精密稱取桑辛素、甘草酸�、異甘草素、桑皮苷A����、地骨皮乙素����、芹糖甘草苷���、甘草素�����、異甘草苷����、甘草苷��、綠原酸�、甘草酸銨、東莨菪內(nèi)酯���、大黃素對照品適量����,用甲醇配制成質(zhì)量濃度分別為30���、82��、53����、65���、60�����、20����、95�、100、77����、85、80�����、165�、38μg/mL的對照品溶液��。
1.3.2 供試品溶液
按照《小兒藥證直訣》中瀉白散配比��,取適量炒桑白皮���、地骨皮、炙甘草��,粉碎�,過孔徑為(250±9.9)μm(65目)篩,混合均勻�����,制成試驗用瀉白散粉末�。精密稱取1.00g瀉白散粉末,加水至30mL�����,回流提取1h�,放冷,補(bǔ)重�,以3000r/min離心5min,過濾上清液,取續(xù)濾液10mL��,按照體積比為1:1加入甲醇�,混勻,靜置過夜��,過濾上清液��,濾液經(jīng)0.45μm微孔濾膜濾過���,得瀉白散供試品溶液,備用���。同法分別制備不同批次的瀉白散�����、炒桑白皮�����、地骨皮�、炙甘草供試品溶液�。
1.4 實驗方法
采用高效液相色譜法,按1.2色譜條件測定瀉白散及其3種單味藥飲片樣品溶液,得到各批瀉白散����、單味藥高效液相色譜指紋圖譜,計算各批瀉白散樣品之間���、單味藥樣品之間���、對照品與瀉白散樣品之間指紋圖譜的相似度,對瀉白散中的共有色譜峰進(jìn)行指認(rèn)�。
2 結(jié)果與討論
2.1 色譜條件優(yōu)化
分別考察了甲醇-甲酸水溶液、甲醇-磷酸水溶液�����、乙腈-甲酸水溶液��、乙腈-磷酸水溶液系統(tǒng)對測定結(jié)果的影響��,結(jié)果發(fā)現(xiàn)乙腈-0.1%甲酸水溶液條件下色譜峰分離度較好��,基線穩(wěn)定����,故選擇乙腈-0.1%甲酸水溶液進(jìn)行優(yōu)化梯度洗脫條件����;分別比較60�、100、120�����、150min的檢測時間對測定結(jié)果的影響��,結(jié)果發(fā)現(xiàn)120����、150min均可檢測出瀉白散中所有的色譜峰,為節(jié)省時間��,選擇檢測時間為120min���;采用DAD-UV檢測器對200~400nm檢測波長進(jìn)行考察,發(fā)現(xiàn)在254nm波長下�,色譜峰最多,瀉白散信息量最大����,最終選擇檢測波長為254nm�����。
2.2 料液比的優(yōu)化
《小兒藥證直訣》記載瀉白散制法及用法“上銼散�,入粳米一撮�,水二小盞,煎七分����,食前服”,依據(jù)《浙江省中藥炮制規(guī)范》(2015年版)確定炒桑白皮工藝為取桑白皮飲片���,照清炒法炒至表面微黃���,微具焦斑時,取出攤涼���。參考王彥帥等確定的經(jīng)典名方瀉白散物質(zhì)基準(zhǔn)制備工藝:取炒桑白皮3.93g���,焙地骨皮3.93g,炙甘草0.39g���,粳米1.50g���,加水360mL煎煮���,料液比約為1:37。遵循古代經(jīng)方制法�����、煎煮原則��,分別將炒桑白皮����、地骨皮、炙甘草粉碎����,過孔徑為(250±9.9)μm篩,進(jìn)行加熱回流提取��,最終確定料液比為1:30��,較為接近古代煎煮工藝�����。
2.3 方法學(xué)驗證
2.3.1 精密度試驗
取桑皮苷A對照品溶液�,在1.2色譜條件下連續(xù)進(jìn)樣6次,記錄色譜峰面積�����。結(jié)果顯示�����,測定結(jié)果的相對標(biāo)準(zhǔn)偏差為0.21%�,表明儀器精密度良好。
2.3.2 穩(wěn)定性試驗
取瀉白散組方X10樣品�����,按1.3.2制備樣品溶液��,在1.2色譜條件下����,分別在0、2����、4����、8����、12、24h進(jìn)樣6次����,記錄色譜峰面積。結(jié)果顯示����,色譜峰面積測定結(jié)果的相對標(biāo)準(zhǔn)偏差為0.25%~1.02%,其中各主要色譜峰保留時間無明顯變化�,測定結(jié)果的相對標(biāo)準(zhǔn)偏差為0.01%~1.21%,表明樣品穩(wěn)定性良好�。
2.3.3 重復(fù)性試驗
取瀉白散組方X10樣品,按1.3.2制備樣品溶液6份�,在1.2色譜條件下各進(jìn)1次,記錄色譜峰面積��。結(jié)果顯示���,色譜峰面積測定結(jié)果的相對標(biāo)準(zhǔn)偏差為0.25%~2.85%�����,其中各主要色譜峰的保留時間無明顯變化����,測定結(jié)果的相對標(biāo)準(zhǔn)偏差為0.13%~1.56%�����,表明該方法的重復(fù)性良好���。
2.4 HPLC指紋圖譜的建立
2.4.1 炒桑白皮指紋圖譜測定
13批全國不同飲片企業(yè)市售桑白皮炒制品(S1~S13)按照1.3.2制備供試品溶液��,在1.2色譜條件下測定�����,將色譜圖導(dǎo)入中藥指紋圖譜相似度評價系統(tǒng)軟件(2004A版)�,HPLC指紋圖譜匹配結(jié)果見圖1�。由圖1可知,13個批次之間的相似度為0.024~0.975���,差別非常大�,炒桑白皮樣品S8中,各成分的含量非常低�,雖然外觀性狀符合炒桑白皮正品特征,但內(nèi)在質(zhì)量非常差����,推測該樣品可能存放時間太久所致。除S8炒桑白皮樣品外�����,其它12批炒桑白皮僅有7個共有峰�����。

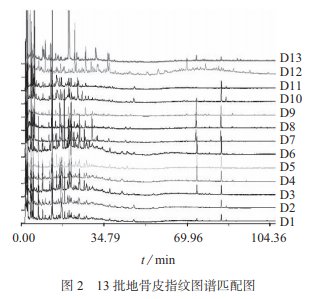
2.4.2 地骨皮指紋圖譜
將13批全國不同飲片企業(yè)市售地骨皮(D1~D13)按照1.3.2制備供試品溶液����,在1.2色譜條件下測定,將色譜圖導(dǎo)入中藥指紋圖譜相似度評價系統(tǒng)軟件(2004A版)����,HPLC指紋圖譜匹配結(jié)果見圖2。由圖2可知���,13個批次之間的相似度為0.061~0.983��,差別非常大�����,地骨皮樣品D11��、D12��、D13與其它樣品的相似度非常低����,D12�����、D13色譜峰數(shù)量及分布與其它地骨皮樣品差別較大�����,雖然外觀性狀均符合地骨皮正品特征��,但內(nèi)在質(zhì)量差異較大�����。13批地骨皮僅有4個共有峰。
聲明:本文所用圖片�����、文字來源《化學(xué)分析計量》���,版權(quán)歸原作者所有�。如涉及作品內(nèi)容�����、版權(quán)等問題����,請與本網(wǎng)聯(lián)系
相關(guān)鏈接:桑白皮,甘草酸�,甲酸,甘草苷



















登錄后才可以評論